
. Introduction
210Pb is a natural geogenic radioisotope that represents part of the uranium decay series and originates from the decay of 226Ra, which is found in most soils and rocks and produces short-lived gaseous 222Rn as its daughter. Most of this 222Rn decays to 210Pb within the soil, producing supported 210Pb, which is essentially in equilibrium with the parent 226Ra. However, some of the 222Rn diffuses upwards into the atmosphere, where it decays to 210Pb (Mabit et al., 2008; Barlas, Simsek and Cagatay, 2014). This 210Pb is deposited as fallout with aerosol particles by washout and sedimentation and commonly termed unsupported or excess (210Pbex) to distinguish it from the supported 210Pb in the soil. Goldberg (1963) was the first who outlined the 210Pb geochronology, Krishnaswami et al. (1971) applied it to the dating of lacustrine sediments and Koide et al. (1972) later applied it to marine sediments. 210Pb excess is extensively used for dating sediments in paleoenvironmental studies (Appleby and Oldfield, 1978; Jia et al., 2006; Zaborska et al., 2007; Kim et al., 2008; Begy et al., 2011).
Depending on different physical and chemical principles, several analytical techniques are available for the measurement of 210Pb. There are several methods for analysing the total activity of 210Pb in the environmental samples based on alpha, beta and gamma spectrometry (Lehto and Hou, 2011; Jia and Torri, 2007). They differ concerning the reachable detection limit, selectivity, analytical error, reproducibility and stability against different chemical composition and levels of the natural radionuclides (Ebaid and Khater, 2006).
Gamma ray spectrometry is a direct counting method of the low-energy (46.5 keV) photon emitted during the decay of 210Pb. However, there are some challenges for biological and environmental samples (Jia et al., 2006). The 46.5 keV photons are emitted in 4% of 210Pb decays. The self-absorption of 46.5 keV line strongly depends on the chemical composition of the sample. Therefore, the high correction factors with associated high relative uncertainties are unavoidable. The minimum detectable activity (MDA) of this technique is higher than that required for the most environmental applications, when maximum sensitivity and accuracy are required, the chemical separation method still remains the first choice.
Alpha and beta techniques for determination of total 210Pb after radiochemical separation are more sensitive and can provide improved analytical precision (Mabit et al., 2008). The main advantage is that the evaluation of 210Pb through the determination of 210Po using alpha spectrometry and 210Bi by beta particles spectrometry has a much lower limit of detection in comparison with gamma spectrometry and requires only gram-size samples (Barlas Simsek and Cagatay, 2014; Zaborska at al, 2007). On the other hand, methods involving chemical separation steps require secular equilibrium to be established between 210Pb and 210Po or 210Bi. This introduces long delays between separation and the counting time (Villa et al., 2005, 2007; Jia and Torri, 2007); as a result these methods become more laborious and expensive compared to gamma ray spectrometry (Mabit et al., 2008).
Determination of 210Pb via its beta emitting daughter 210Bi using liquid scintillation counting (LSC) technique could be an alternative to alpha spectrometry, as it eliminates long wait for secular equilibrium, and provides a comparable detection limit with alpha spectrometry (Villa et al., 2007). The radiochemical Pb separation procedure by precipitation has been recommended by many authors (Goldberg, 1963; Polikarpov, 1966; Dušauskienė-Duž, 1997; Jia et al., 2001; Mažeika et al., 2004) due to its quite easy operation and short preparation time. Using this technique, Pb is separated by precipitation as lead sulphide (PbS) or lead sulphate (PbSO4) and measured with liquid scintillation or proportional counter. Before precipitation, interfering bismuth is separated from the solution by an anion exchange resin column. The advantage made with the help of 210Bi is that the measurement does not need to be made immediately after the separation of 210Pb (Lechto and Hou, 2011). This method has a wide applicability for measuring low level 210Pb concentrations in most environmental samples (Jia and Torri, 2007).
The most commonly used models for 210Pb chronology are based on radioactive decay equations for converting 210Pbex data from a sediment profile into a sediment chronology and include: the Constant Flux: Constant Sedimentation (CF:CS), Constant Rate of Supply (CRS) and Constant Initial Concentration (CIC). Descriptions of the models and the associated equations have been reported in (Krishnaswamy et al., 1971; Pennington et al., 1976 and Appleby, 2001). Model selection depends on environmental conditions, sediment processes, such as bioturbation, erosion, deposition.
The CIC model is appropriate when initial activity of 210Pbex is constant and there is no mixing of surface sediments (MacKenzie et al., 2011 and Mabit et al., 2014). The CRS model assumes that the supply of 210Pbex to the accreting material is constant in time (Appleby and Olfield, 1978), the initial 210Pb concentration in the sediments is variable, and the influx rate of sediment is variable (Goldberg, 1963 and Appleby and Oldfield, 1978). When 210Pbex fluxes and sediment mass accumulation rate (MAR) are both constant, the CIC and CRS models converge to CF:CS model and the 210Pbex concentration follows an exponential decrease with mass depth (Mabit et al., 2014).
210Pb dating depends on the accurate determination of the level of unsupported 210Pb in a series of sediment samples, what is quite problematic because of sampling and analytical limitations (MacKenzie et al., 2011). Loss of surface material during sediment sampling can generate inaccuracies in chronology as described by Farmer et al. (2006). The validation of chronology based on 210Pb dating models for the second half of the 20th century is often achieved by the use of artificial fallout radionuclides (e.g., 137Cs and 241Am) from the atmospheric testing of nuclear weapons and/or the Chernobyl accident as independent chronostratigraphic markers (O’Reilly et al., 2011).
The aim of the present study was, firstly, to compare two nuclear analytical techniques for 210Pb analysis in sediment samples using LSC and gamma-ray spectrometer with well type HPGE detector and, secondly, to perform 210Pb dating of sediment cores attributed to two lakes located in the Baltic Uplands.
. Materials and methods
The study was carried out on sediment cores from two lakes (Fig. 1) located in the Baltic Uplands. Lake Karackiai is a little kettle-hole type lake located in eastern part of Lithuania, near the border with Belarus. The lake covers area of 17 ha with maximal depth of 10 m. Forests occupy a prevailing part of Lake Karackiai catchment. Lake Rajgrod is located in a glacial tunnel valley in the northeastern part of Poland. The lake covers an area of 1500 ha, its greatest depth is 52 m and average is 9.4 m (Krzywicki et al., 2007). The sampling area was attributed to the eastern part of lake. The Lake Karackiai is insignificantly impacted by anthropogenic activity. The Lake Rajgrod is impacted by agriculture, also there are water level regulation by dam constructed on out flowing stream (in 50s XX c.). Based on visual evaluation Lake Rajgrod is more eutrophic compared with Lake Karackiai.
Sampling
The sediment cores were collected using a Kajak gravity corer (producer KC Denmark A/S Research Equipment) from boat in 2010 and 2013. In Lake Karackiai the sampling point was attributed to the deepest central depression (mean depth of 8 m). In Lake Rajgrod the sampling point was attributed to the deepest depression (mean depth of 19 m) of the eastern part of lake.
The sediment cores, of length 30 cm (Lake Karackiai) and 45 cm (Lake Rajgrod), were sectioned in situ, into 1 cm and 2.5 cm slices, respectively. The sliced sediment material was sealed in plastic boxes and transported to the laboratory.
Dry mass was determined after drying samples at 105°C, and then together with wet sample volume were used to calculate the dry bulk density. The samples were homogenized and stored for later treatment. Firstly, the sediment samples were used to fill the measuring containers (mini beaker of 3 ml) for gamma spectrometry assay to measure 210Pb, 214Pb and 137Cs. After the measuring by gamma spectrometry, the same sediment samples were involved into radiochemical procedures for the determination of the total 210Pb in equilibrium state of system 210Pb – 210Bi by LSC.
Radiochemical procedure
The first step of pre-treatment involved the drying of the sample at 105°C temperature, crushing, grinding and sieving through 2 mm sieve mesh size. For ashing, 10 g of the dried sample was taken and ashed at 550°C for 6 hours. Finally, the sample ash was ground and homogenized.
Leaching
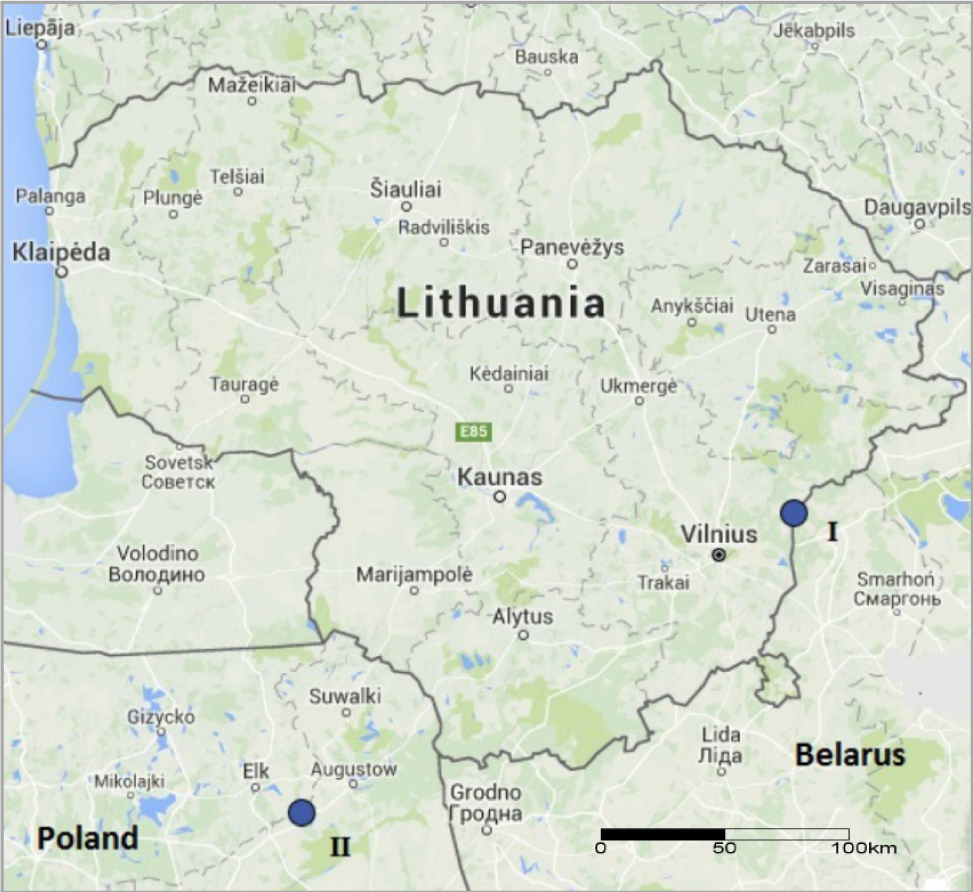
There are used several leaching methods as reported in Jia et al., 2006: (i) leaching with aqua regia; (ii) successive leaching with HNO3 + HF, HClO4 and HCl, etc. To remove nearly all organic matter and silicates from the samples, which can prevent the recovery of 210Pb and decrease the radiochemical yield, samples oxidation and leaching were carried out followed by wet ashing in our study. After the sample pre-treatment, the chemical separation was started by adding to the beaker containing 3 g of the ashed sample 1 ml of stable Pb2+ carrier (30 mg/ml) as the chemical yield tracer. For the further procedure, 5 ml of HNO3 and 15 ml of HCl were added to samples and heated at 250°C to evaporate the solution to dryness. The remaining material was treated with 10 ml of HCl (1M) to finally destroy the organic matter. Solution was evaporated to dryness again. The residue was finally dissolved with 30 ml of 1M HCl, filtered through a 0.1 µm Millipore filter paper, and transferred to a 50 ml volumetric beaker.
Separation
Pb was separated from the leachate solution using anion exchange resin column in chloride form (Eichrom 1 ×8, 100–200 mesh). Ion-exchange column was approx. 130 mm long and 11 mm of inner diameter. The anion exchange resin was sequentially treated with 20 ml of 2M H2SO4, 20 ml of 6M HNO3 and 100 ml of distilled water to remove fine particles as well as other components after each separation procedure.
The scheme of procedure for the 210Pb separation from leachate solution is shown in Fig. 2.
The available leachate solution from the sample was loaded onto an anion exchange resin column preconditioned with 20 ml of 2M HCl at a flow rate 0.5 ml min–1. After rinsing with 10 ml of 1M HCl supernatant was discarded. Under this condition (10 ml of 1M HCl) Bi should be fixed on the resin in contrast to Pb (Happel et al., 2006). From the loaded anion exchange resin column Pb could be separated by elution with 0.05M or 0.5M HCl (Figgins, 1961; Gibson, 1961 and Bhatki, 1977). H2O as the Pb elution liquid to separate Pb from Bi can be used as well (Jia and Torri, 2007). In our study Pb was eluted with 50 ml of H2O at the same flow rate. The separation time of Pb from Bi was measured.
For determination of the radiochemical yield gravimetrically 4 ml of H2SO4 were added to the eluate in order to precipitate lead as sulphate (PbSO4). After evaporation of solution till volume of 20–30 ml the precipitate clearly occurred. Then volumetric beaker with content was cooled and precipitate was filtered through ashless Whatman filter. Filter paper with precipitate was put in a weighed crucible, ashed at 550°C for 4 hours, cooled until constant weight and weighed again to calculate the Pb radiochemical yield based on Pb2+ carrier added.
Dissolution
According to Jia et al. (2006) the PbSO4 precipitate can be dissolved in sufficient volume of 6M ammonium acetate by heating. Nearly in the same way Letho and Hou (2011) recommended to dissolve the lead sulphate in 10 ml of 6M ammonium acetate. Other solutions, including EDTA/NaOH and ammonium citrate, were investigated for PbSO4 dissolution (Kim et al., 2008 and Lozano et al., 2012). In our case, PbSO4 was distributed in 2 ml of 3M ammonium acetate, mixed with 18 ml of scintillation cocktail (Optiphase HiSafe 3, Perkin Elmer) in a 20 ml plastic vial and stored for over 30 days in a refrigerator for consequent LSC assay.
Model samples and radiochemical yield
To test the reproducibility of radiochemical technique, model samples not containing unsupported 210Pb and referred as sets #1, #2 and #3 were taken from deeper layers of sediment core of Lake Rajgrod. Samples were spiked with approx. 0.1 ml of 210Pb tracer solution (specific activity 41.65 Bq/g, uncertainty 1.1%, solution based on Pb(NO3)2 in HNO3, Czech Metrology Institute). Then the radiochemical procedure described above (Section 2 – Radiochemical procedure) has been applied on the same samples. This experiment allowed to evaluate radiochemical yield by two methods – based on added Pb2+ carrier and on 210Pb tracer.
The PbSO4 precipitates from the model samples were alternatively dissolved with: 2 ml of 6M ammonium acetate (#1); 2 ml of 3M ammonium acetate (#2) and 2 ml of 0.5M EDTA/NaOH (#3). 2 ml of resulted solution were mixed with 18 ml of Optiphase HiSafe 3 scintillation cocktail in measuring vial, which was stored for 30 days, afterwards was tested on homogeneity and finally measured by LSC.
For model samples, radiochemical yield by 210Pb tracer was calculated according to formula:
where, Rt – total counting rate (s–1) in 210Pb–210Bi spectrum window, from 30 to 770 channels (for equilibrium state counting rate for whole region are divided by 2); Rb – background counting rate (s–1) in 210Pb–210Bi spectrum window; APb – activity added to calibration sample (Bq); εβ – counting efficiency.
Liquid scintillation counting
Model, blank, standard and unknown samples were measured using a QUANTULUS 1220 low-background scintillation spectrometer in the same assay conditions using a pulse shape analyser (PSA) function to achieve separation of alpha and beta events (Vila et al., 2007). The pulse shape α/β discriminator optimized at PSA level 125 was used to separate possible interferences of alpha emitters present in the sample.
For the blank sample, 1 ml Pb2+ carrier (30 mg/ml) was added to 30 ml of distilled water and then subjected to the same radiochemical procedure as described above (Section 2 – Radiochemical procedure). The counting efficiency for β emitters was evaluated with the 210Pb standard which was prepared by adding 1 ml Pb2+ carrier (30 mg/ml), 1 ml of a standard solution of 210Pb (24.3 Bq/g) to 30 ml of distilled water. The sample then followed the same procedure as for the blank sample.
Assuming that 210Pb and 210Bi after 30 days reached a secular equilibrium, counting efficiency was calculated from the standard sample using total counting rate in the 210Pb -210Bi window by formula:
where, Rt – total counting rate (s–1) in 210Pb–210Bi spectrum window; Rb – background counting rate (s–1) in 210Pb– 210Bi spectrum window; Yg – radiochemical yield.
210Pb activity concentration (Bq/g) in samples was calculated as:
where, Rt – total counting rate (s–1) in 210Pb–210Bi spectrum window; Rb – background counting rate (s–1) in 210Pb–210Bi spectrum window; Yg – radiochemical yield; εβ – counting efficiency; m – mass of the sample (g).
Gamma spectrometry
210Pb, 214Pb and 137Cs activities were determined by gamma spectrometry using HPGe GWL-series detector (detector diameter 54.7 mm, detector length – 67.8 mm, active well depth – 40 mm, well inside diameter – 15.5 mm, total active volume – 126 cm3, absorbing layers of high purity aluminium – 0.5 mm, inactive germanium – 0.3 µm, resolution, FWHM, at 1.33 MeV (60Co) – 2.25 keV, and at 122 keV (57Co) – 1.2 keV). 210Pb activity concentrations were measured via its gamma ray emission at 46.5 keV, 137Cs at 661.7 keV and 214Pb, as a shortlived product of 226Ra, at 295.2 keV and 351.9 keV. GammaVision-32 software was used for activities calculation and quality assurance procedure. The gamma spectrometric system was calibrated for counting efficiency using commercially available multi radionuclides standard sources and reference solution for different densities and filing heights with selected matrix as described in detail (Marčmliomene et al., 2015). The detection limit for the counting time of 200 000 s was about 0.014 Bq for 137Cs, 0.065 Bq for 210Pb and 0.021 Bq for 214Pb, while measurement errors did not exceed 8%, 15% and 20% for 137Cs, 210Pb and 214Pb, respectively. The precision of gamma spectrometric measurements of our laboratory was approved during the comparison exercise organized by Lithuanian Metrology Inspectorate and Center for Physical Sciences and Technology in 2013, and by the STUK – Finish Radiation and Nuclear Safety Authority in 2014.
. Results and discussion
The results of the radiochemical yield (gravimetrical and by 210Pb tracer) obtained from model samples and other methodical features (solubility and homogeneity of measuring form for LSC) are given in Table 1.
Table 1
Comparison of investigated radiochemical procedures.
The radiochemical yield of Pb evaluated by both methods was as follows: 72–77% for gravimetric method and 70–73% for 210Pb tracer method. All three solvents used to dissolve PbSO4 precipitates were suitable. The long-term stability of dissolved PbSO4 mixture with 18 ml scintillation cocktail Optiphase HiSafe 3 was confirmed by clear and homogenous measuring form with 2 ml of 6M ammonium acetate (#1) and 2 ml of 3M ammonium acetate (#2). Whereas PbSO4 dissolved in 2 ml of 0.5 EDTA/NaOH and mixed with 18 ml of scintillation cocktail resulted a precipitate formation on the bottom of the vial after 1 day. The counting efficiency based on standard and blank counting rates in the 210Pb– 210Bi window (30–770 channels) for PSA level 125 varied between 88% and 91%. Based on experimental results (Table 1) 2 ml of 3M ammonium acetate solution was used further in radioanalytical procedure for 210Pb determination in lakes sediment samples by LSC method.
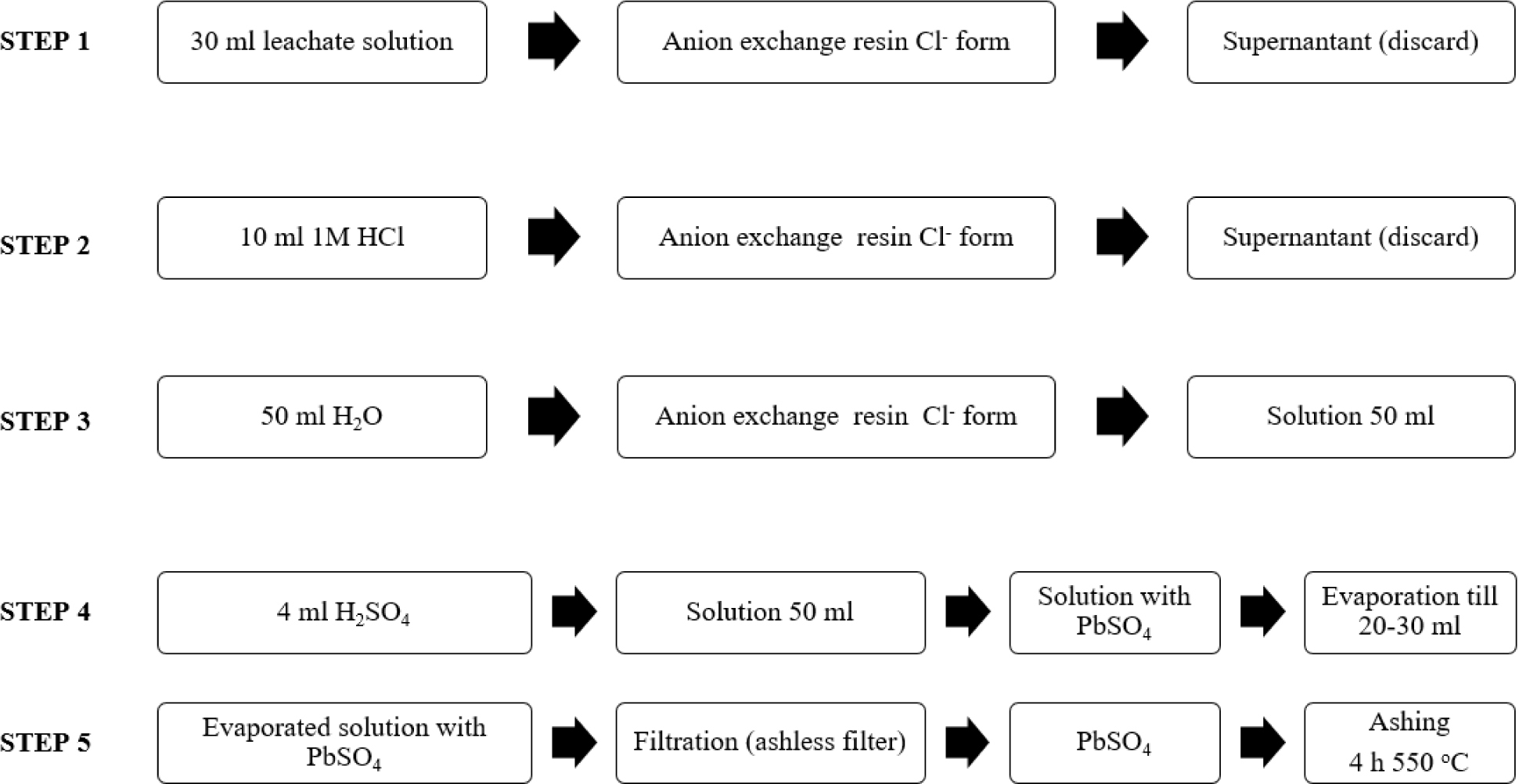
A set of samples from two cores of both lakes was prepared and measured. The data obtained from direct measurements of 210Pb (gamma spectrometry) and from indirect measurements (LSC) was used to compare the results of different measurement techniques. The results of 210Pb activities versus core depth for both methods are shown in Fig. 3 (Lake Karackiai) and Fig. 4 (Lake Rajgrod).
Fig. 3
Distribution of the total 210Pb measured by two methods versus core depth of Lake Karackiai (Lithuania).
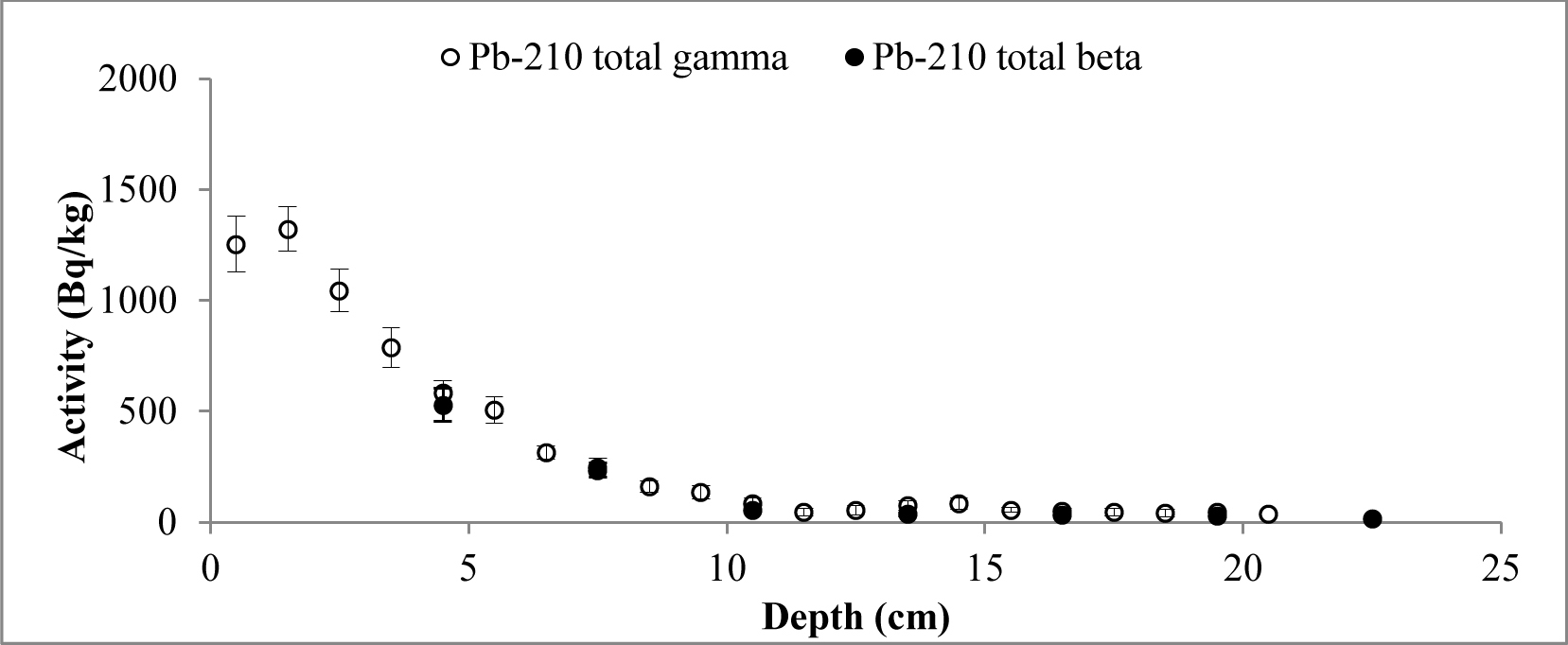
Fig. 4
Distribution of the total 210Pb measured by two methods versus core depth of Lake Rajgrod (Poland).
It is evident that, except for a few samples from Lake Rajgrod core (Fig. 4), the210Pb activity concentrations obtained using both methods are similar within the range of uncertainties. The observed shift between results for particular samples are likely to be related to an incomplete homogenization of those samples. In fact, analysis of the sediments between three methods, alpha and gamma spectrometry, and LSC (Villa et al., 2007) has shown the similar discrepancies, which can be attributed to insufficient processing as well. Also, a trend of higher activities of 210Pb measured by gamma compared to LSC for the upper depth intervals of Lake Rajgrod core can be observed. The relative uncertainty associated with gamma spectrometry measurements of 210Pb is quite large for samples of Lake Rajrod (Fig. 4). At low 210Pb activity (20 Bq kg–1) uncertainly associated with gamma spectrometry measurements can reach ±70%, although for higher activities (>60 Bq kg–1) uncertainties become acceptable (±20%) and when activities exceed 110 Bq kg–1, uncertainty levels are close to those associated with LSC measurements (±15%). A similar systematic uncertainty between measuring techniques were also noticed by Tanner et al. (2000), Zaborska et al. (2007) and Mabit et al. (2008).
The 210Pb excess (unsupported), which is used for age calculation, was determined subtracting supported 210Pb (214Pb) from total 210Pb. The total 210Pb activities were not determined for each sediment slice by LSC. Further for age calculation 210Pbex activity extrapolation from the exponential function was applied for comparison of two age data sets.
Activity of unsupported 210Pb decreases with depth for the both cores, however some differences from the exponential function can be observed for surface layers. 210Pbex profile for Lake Karackiai exhibits surface mixing with depth of approx. 2 cm (Fig. 5), while mixing at Lake Rajgrod profile was obtained to approx. 11 cm depth (Fig. 6). The 210Pb equilibrium depth reached 12 cm for Lake Karackiai core (Fig. 5) and 30 cm for Lake Rajgrod core (Fig. 6). Figs. 5 and 6 illustrate only the 210Pbex results which were used for age-depth model.
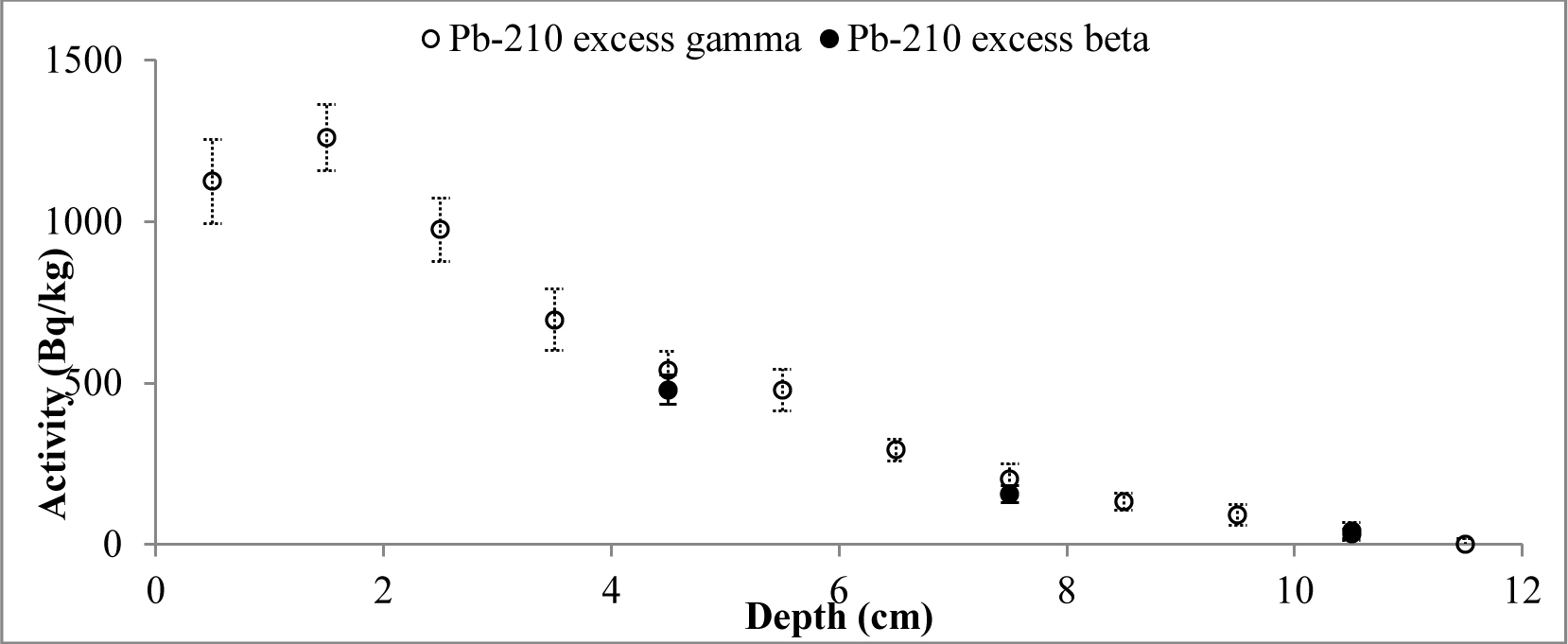
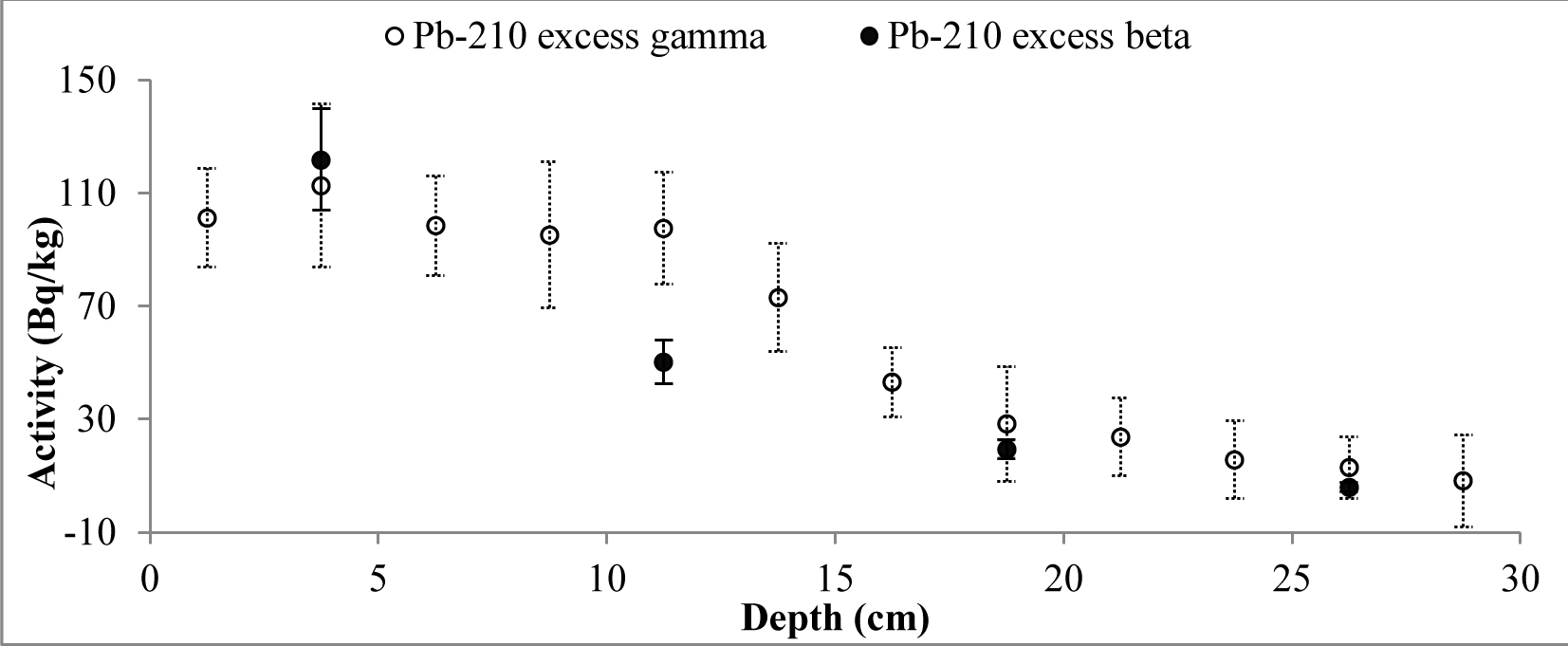
The inventories of 210Pbex (Bq/m2) were calculated based on cumulative sum of the individual 210Pbex activity values (Bq/kg) determined by both beta and gamma methods over the entire profiles, multiplied by the mass depth of each sediment layer (g/cm2). The results of both sites included in this study were found to be comparable. In fact, the calculated 210Pbex inventories results derived by beta and gamma methods for Lake Karackiai are 2579 and 2783 Bq/m2 while for Lake Rajgrod are 1918 Bq/m2 and 2650 Bq/m2, respectively.
The CRS model could be used to derive 210Pb chronology of sediments as a 210Pbex activity with depth is highly variable (Goldberg, 1963; Appleby and Oldfield, 1978; Barlas Simsek and Cagatay, 2014). Therefore, the sediment age, sedimentation rate (SR) and mass accumulation rate (MAR) values for studied cores were determined by the CRS model (Appleby et al., 1991). The 210Pb age-depth relationships for CRS model of two cores are shown in Figs. 7 and 8.
Fig. 7
CRS model 210Pb dates versus depth for Lake Karackiai core. There are also shown 137Cs activity changes versus depth and dates derived from 210Pb CRS model.
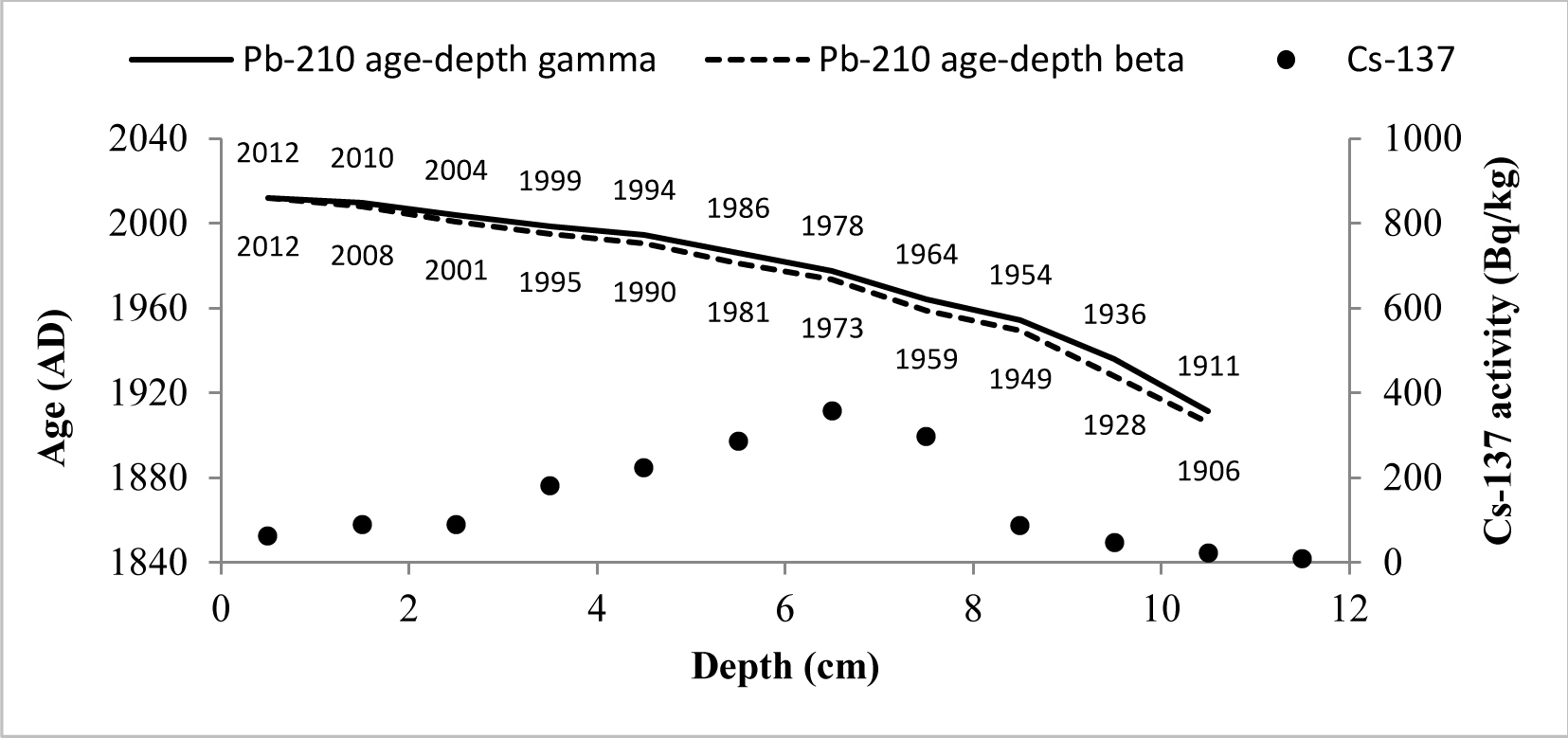
Fig. 8
CRS model 210Pb dates versus depth for Lake Rajgrod core. There are also shown 137Cs activity changes versus depth and dates derived from 210Pb CRS model.
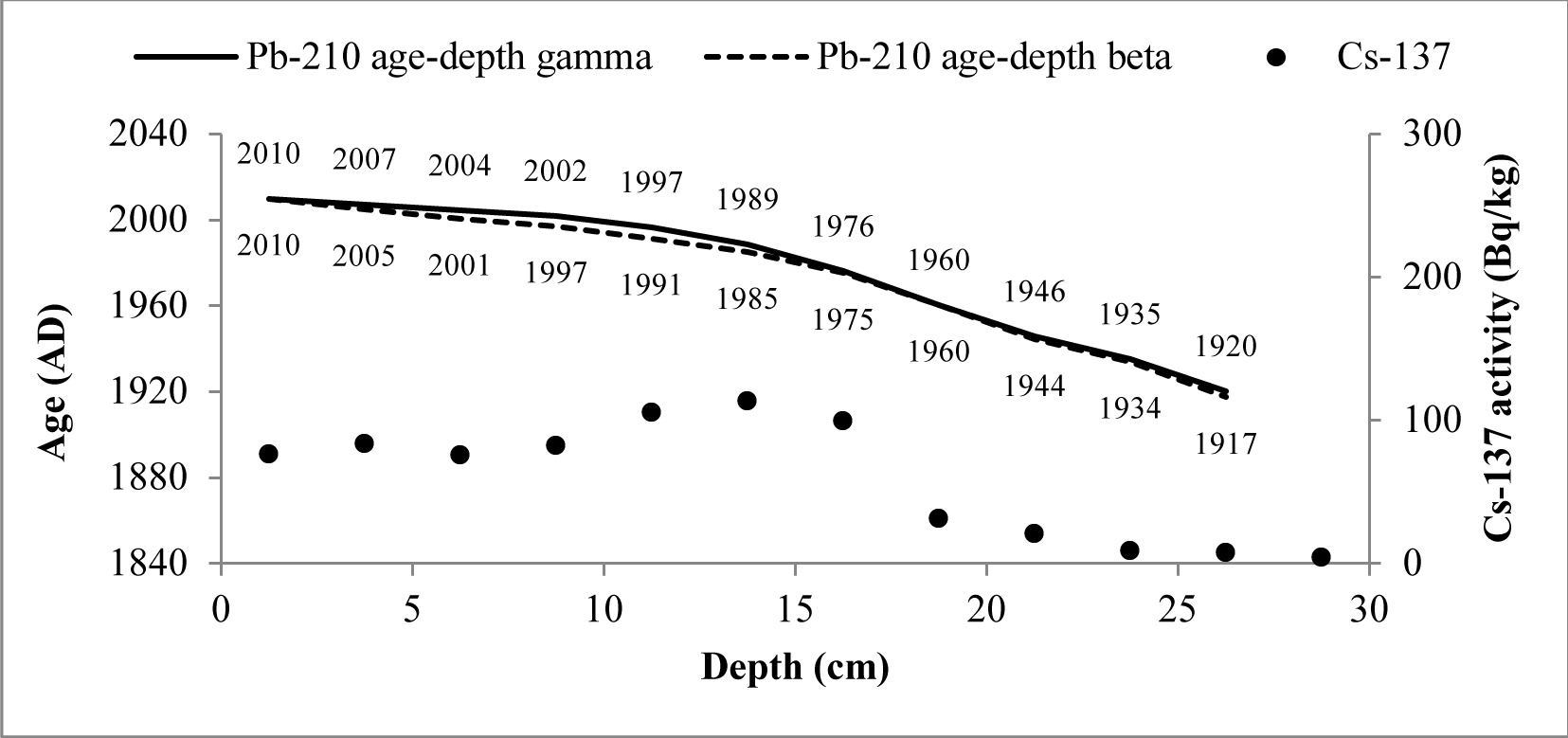
The artificial fallout radionuclide 137Cs data representing independent chronomarker are included into those graphs for 210Pb chronology validation (Pennington et al, 1973; Smith, 2001 and Walling et al, 2002). 137Cs inventory in core of Lake Karackiai was estimated 1470 Bq/m2, while in core of Lake Rajgrod was estimated 3100 Bq/m2. The Lake Rajgrod 137Cs inventory data significantly high imply the Chernobyl accident fallout impact on catchment.
The 137Cs peak was found at depth of 6–7 cm (359±19 Bq/kg) in core of Lake Karackiai and at depth of 12–15 cm (114±6 Bq/kg) in core of Lake Rajgrod and can be attributed to Chernobyl accident. Those sediment layers were dated by 210Pb CRS model as 1973–1978 and 1985–1989, respectively. Some discrepancies between 210Pb CRS model dates and 137Cs peak position could be explained as 137Cs penetration to greater depth than expected from diffusion (Mabit et al., 2014). It can be influenced by combination of physical and biological mixing of newly accumulated sediments (O’Reilly et al., 2011), as well as by postdepositional migration in sediments (Appleby et al., 1991).
The comparing of sedimentation parameters evaluated by 210Pbex data obtained by two analytical methods is presented in Table 2.
Table 2
Sediment mass accumulation and sedimentations rates (mean values) based on 210Pbex evaluated by gamma and beta methods.
| Core | Mass accumulation rate (g/cm2/yr) | Sedimentation rate (cm/yr) | ||
|---|---|---|---|---|
| gamma method | gamma method | beta method | beta method | |
| Lake Karackiai | 0.009 | 0.008 | 0.14 | 0.12 |
| Lake Rajgrod | 0.060 | 0.055 | 0.41 | 0.33 |
From 210Pb chronology derived mean values of sediment mass accumulation rate (MAR) were following: 0.008 g/cm2/yr based on beta method and 0.009 g/cm2/yr based on gamma method for Lake Karackiai; 0.055 g/cm2/yr based on beta method and 0.060 g/cm2/yr based on gamma method for Lake Rajgrod. The mean values of linear sedimentation rate were: 0.14 cm/yr using gamma data and 0.12 cm/yr using beta data for Lake Karackiai; 0.41 cm/yr using gamma data and 0.33 cm/yr using beta data for Lake Rajgrod. The differences in accumulation rate in these two lakes might be due to the damming of the Lake Rajgrod in mid 50s of XX c. The water level of the lake was raised some 1–1.5 m. After the II world war the town developed and due to the lack of environmental awareness the lake waters were polluted what increased the rate sedimentation (Krzywicki et al., 2007).
. Conclusions
210Pb activity concentration in lacustrine sediments was determined using two nuclear analytical techniques including beta LSC and HPGe gamma spectrometry. Both techniques in the range of uncertainties gave similar 137Cs, 210Pb and 226Pb measurement results. The large relative uncertainty associated with gamma analysis of samples with low 210Pb activity makes beta LSC technique attractive in some circumstances even if this method is labour consuming. The values of 210Pbex inventory derived using data from beta and gamma measurements were found to be similar with for Lake Karackiai (2579 and 2783 Bq/m2) and slightly variable for Lake Rajgrod (1918 and 2650 Bq/m2). Among two lakes more eutrophic Lake Rajgrod exhibited the higher sediment mass accumulation rate (0.055 and 0.060 g/cm2/yr) compared to less eutrophic Lake Karackiai (0.008 and 0.009 g/cm2/yr). The 210Pb chronologies with some limitations were validated using artificial fallout radionuclide 137Cs as independent chronomarker.